Lucerastat is an investigational, oral inhibitor of glucosylceramide synthase, offering a potential new treatment approach for all patients living with Fabry disease, irrespective of the mutation type of the GLA gene.
Lucerastat
Lucerastat is an oral inhibitor of glucosylceramide synthase
Fabry disease is a rare, X-linked lysosomal storage disorder caused by mutations in the GLA gene that results in the absence or markedly reduced activity of the enzyme α-galactosidase A (α-GalA). α-GalA normally breaks down a fatty product known as globotriaosylceramide (Gb3) in the cells of the body. Deficiency over time results in an accumulation of Gb3 deposits throughout the body, leading to multisystem disease, mainly affecting the kidneys, heart, and nervous system.
The disease manifests in two main phenotypes: classic Fabry disease, typically presenting in childhood with severe, multisystemic involvement, and late-onset Fabry disease, which may emerge in adulthood with predominant cardiac or renal symptoms. Fabry disease affect a patient’s life expectancy and quality of life. Due to its variable presentation and non-specific symptoms, Fabry disease is frequently underdiagnosed or misdiagnosed, leading to delays in treatment and increased risk of irreversible organ damage.
As the gene responsible for Fabry disease is found on the X chromosome (of which males have one, and females two), males with deleterious mutations have little or no residual alpha-GalA activity. Therefore, these male patients with Fabry disease experience a wider spectrum of symptoms, and in some cases, a greater severity. It is now widely accepted that women with Fabry disease are heterogeneous with respect to disease severity and may sometimes also develop life threatening complications of the disorder. Up to 70% of female carriers develop Fabry related symptoms at some point in their life.
According to DelveInsight, approximately 16,000 patients were diagnosed with Fabry disease across the seven major markets (US, EU4, UK, Japan) in 2020, a number expected to increase to roughly 21,000 by 2034. The Fabry disease market is projected to reach around USD 4 billion by 2034, underscoring the continued demand for innovative, patient-focused therapies.

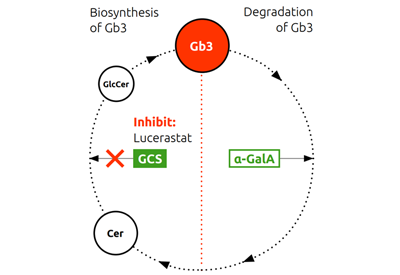
The normal biosynthesis and degradation of Gb3 is shown schematically in the Figure below. In patients with Fabry disease, deficiency or dysfunction of the enzyme alpha Gal A leads to abnormal accumulation of Gb3, which in turn causes the symptoms of Fabry disease.
Current treatment options include enzyme replacement therapies (ERTs) and oral chaperone therapy for patients with amenable mutations. However, these therapies have limitations, including intravenous administration, immunogenicity, and mutation-specific efficacy. There remains a significant unmet need for a well-tolerated, oral, disease-modifying therapy that can be used regardless of genotype or prior treatment history.
In contrast, lucerastat, an oral inhibitor of glucosylceramide synthase (GCS), reduces the substrate which forms Gb3. Substrate reduction therapy (SRT) decreases the build-up and is thought to subsequently reduce the Gb3 load in patients with Fabry disease. Since this mechanism is independent of alpha Gal A deficiency or dysfunction, it should not be limited to specific mutations of the GLA gene.
The Gb3 cycle

Abbreviations: α-GalA, α-galactosidase A; Cer, ceramide; Gb3, globotriaosylceramide; GCS, glucosylceramide synthase; GlcCer, glucosylceramide; Sph, sphingosine
Lucerastat is an investigational, oral substrate reduction therapy designed to treat Fabry disease independently of α-Gal A activity, GLA mutation status, or prior enzyme replacement therapy (ERT). It acts by inhibiting glucosylceramide synthase, thereby reducing the synthesis of glycosphingolipids, including globotriaosylceramide (Gb3), which accumulate due to deficient α-galactosidase A activity in Fabry disease.
Preclinical studies demonstrated that lucerastat is a highly soluble and bioavailable small molecule capable of penetrating key tissues affected by Fabry disease – including the kidneys, liver, and dorsal root ganglia – where it effectively reduces substrate accumulation. Clinical pharmacology studies confirmed lucerastat’s favorable pharmacokinetic profile, characterized by rapid absorption, predictable elimination, and no evidence of saturation, supporting consistent exposure across dosing regimens.
Across Phase 1 studies, lucerastat was well tolerated at doses up to 4000 mg, with no dose-limiting toxicities and a safety profile unaffected by concomitant medications. In a 12-week exploratory study in adult Fabry patients receiving ERT, lucerastat 1000 mg twice daily led to a rapid and sustained reduction in plasma Gb3 and related biomarkers, confirming its mechanism of action and potential for fast-onset substrate reduction.
The recently published Phase 3 MODIFY study and its long-term extension further support lucerastat’s disease-modifying potential. While the primary endpoint of neuropathic pain reduction was not met, lucerastat demonstrated robust and sustained biomarker reductions and a promising renal signal, with a slower rate of eGFR decline in patients with impaired kidney function. These findings suggest lucerastat may offer long-term organ protection and broaden therapeutic options for Fabry patients, especially those underserved by current treatments.
MODIFY was a multicenter, double-blind, randomized, placebo-controlled, Phase 3 study evaluating lucerastat as an oral monotherapy for adults with Fabry disease. A total of 118 patients from 14 countries were randomized 2:1 to lucerastat or placebo. Upon completion of the double-blind period, 107 patients entered a long-term OLE assessing safety, tolerability, and renal outcomes. Primary results from MODIFY and the 12-month interim OLE analysis have been published in Nature Communications, (“Lucerastat, an oral therapy for Fabry disease: Results from a pivotal phase 3 study and its open-label extension”, January 2026).
While the MODIFY study did not meet its primary endpoint of reducing neuropathic pain over six months, lucerastat demonstrated a robust pharmacodynamic effect, significantly reducing plasma and urinary Gb3 levels compared to placebo. These reductions were sustained over time in the OLE, with patients switching from placebo to lucerastat showing similar biomarker reductions.
More importantly, as presented at WORLDSymposium™ 2026, an interim analysis of the OLE, where ongoing patients had been treated with lucerastat for at least 42 months – some exceeding six years of continuous therapy – revealed a notable shift in renal function trajectory. Treatment with lucerastat was associated with a reduction in the rate of eGFR decline as compared to eGFR slope observed in the 2 years preceding their enrollment in MODIFY (eGFR slope, historical: -3.50; Lucerastat: -1.64). Lucerastat treatment was also associated with a particularly marked attenuation of kidney function loss in patients with severe disease course, such as classic males with Fabry disease (historical: -4.32; Lucerastat: -2.05), patients with impaired renal function at baseline (eGFR <90 mL/min/1.73m2) (historical: -6.18; Lucerastat: -2.49), Anti-Drug Antibody (ADA+) positive patients (historical: -7.75; Lucerastat: -3.43). The effect was independent of the gene variants’ amenability to migalastat. Lucerastat also led to a 51% decrease in plasma Gb3 levels and was well-tolerated with long-term treatment. As the next steps for lucerastat are underway, the OLE study will be concluded. To ensure continuity of care for participants still receiving lucerastat at study closure, a post-trial access program is being established. (Wallace E., et al. Mol Genet Metab. 2026;147(2):109659)
In addition, the positive impact on kidney function was supported by a kidney biopsy sub-study of six male adult patients with classic Fabry disease who had received lucerastat monotherapy for a median treatment period of 56 months. The study evaluated the number of kidney Gb3 inclusions using established quantitative (Barisoni Lipid Inclusion Scoring System (BLISS)) and semi-quantitative (Fabrazyme Scoring System (FSS)) methodologies. At enrollment in MODIFY, 4 participants were ERT naïve or pseudo-naïve and 2 switched from ERT. Median kidney Gb3 BLISS score was 1.7 (range: 0.7–4.5; mean (SD): 2.41 (1.59)). Mean FSS scores were 0 indicative of “no or trace” accumulation (5/6 participants) or 1 indicative of mild accumulation (1/6). The 2 patients who switched from ERT had BLISS scores of 1.6-1.8 and a FSS score of 0. The results indicate that long-term treatment with lucerastat monotherapy was associated with low-to-no levels of kidney Gb3 inclusions. (Germain DP., et al. Mol Genet Metab. 2026;147(2):109423)
Lucerastat was well tolerated. No clinically meaningful changes in vital signs or ECGs or marked laboratory abnormalities were observed. Two patients in each group (lucerastat 2.5%; placebo 5.4%) discontinued treatment due to adverse events. Serious adverse events were reported in 5 patients (6.3%) in the lucerastat group and in 1 patient (2.7%) in the placebo group.
Lucerastat for Fabry disease has received orphan drug designation in the US and the EU and is under review in Japan.
Current status
Idorsia has aligned with the US FDA on a streamlined registration strategy comprising two complementary clinical trials designed to definitively characterize lucerastat’s renal effects. This program is also in line with the feedback received from European Medicines Agency.
Pivotal kidney biopsy study (n=16)
- Adult males with Fabry disease, treatment‑naïve or pseudo‑naïve;
- Designed to show a decrease from baseline in renal Gb3 burden after 18 months of treatment.
Renal function comparative study (n ≈ 74)
- Adult patients with Fabry disease;
- Assessment of lucerastat versus established enzyme replacement therapies (agalsidase beta, pegunigalsidase alfa);
- Designed to reinforce lucerastat as the first oral therapy suitable for all patients with Fabry disease, irrespective of their mutation type.
The program is expected to support a potential regulatory filing as early as 2029.
Milestones
2026 New data and evaluation of long-term treatment with lucerastat presented at
WORLDSymposium™ 2026
2021 Phase 3 open label extension study continues
2021 Phase 3 study completed – primary endpoint not met
2018 Phase 3 study initiated
2016 Phase 1b study completed
Key scientific literature
-
Nordbeck P., et al. Nature Communications, 10 January 2026 (online ahead of print).
-
Guérard N., et al. Clin Pharmacol Ther. 2018; 103(4):703-11.
- Welford RWD., et al. Hum Mol Genet 2018; 27(19): 3392-3403.